

Authors: Candela Iglesia, Mathieu Ringeard, Francesca Di Nunzio, Jeliette Fernande, Raphael Gaudin, Philippe Souque, Pierre Charneau, Nathalie Arhel
The human immunodeficiency virus type 1 (HIV-1) central DNA Flap is generated during reverse transcription as a result of (+) strand initiation at the central polypurine tract (cPPT) and termination after a ca. 100 bp strand displacement at the central termination sequence (CTS). The central DNA Flap is a determinant of HIV-1 nuclear import, however, neither cPPT nor CTS mutations entirely abolish nuclear import and infection. Therefore, to determine whether or not the DNA Flap is essential for HIV-1 nuclear import, we generated double mutant (DM) viruses, combining cPPT and CTS mutations to abolish DNA Flap formation.
The combination of cPPT and CTS mutations reduced the proportion of viruses forming the central DNA Flap at the end of reverse transcription and further decreased virus infectivity in one-cycle titration assays. The most affected DM viruses were unable to establish a spreading infection in the highly permissive MT4 cell line, nor in human primary peripheral blood mononuclear cells (PBMCs), indicating that the DNA Flap is required for virus replication. Surprisingly, we found that DM viruses still maintained residual nuclear import levels, amounting to 5-15% of wild-type virus, as assessed by viral DNA circle quantification. Alu-PCR quantification of integrated viral genome also indicated 5-10% residual integration levels compared to wild-type virus.
This work establishes that the central DNA Flap is required for HIV-1 spreading infection but points to a residual DNA Flap independent nuclear import, whose functional significance remains unclear since it is not sufficient to support viral replication.
During infection, the reverse transcriptase (RT) of retroviruses converts the (+) strand RNA genome into double-stranded DNA prior to nuclear import. Similar to all DNA polymerases, the retroviral RT requires the 3' OH of a primer to initiate polymerisation. The primer used for (-) strand synthesis is a cellular tRNA already present in the retroviral particles prior to infection and anneals to the 5' region of the genome at the primer binding site (PBS). The primer used for (+) strand synthesis is a polypurine tract (PPT) present in the 3' region of the RNA genome that resists RNase H degradation concomitant with (-) strand synthesis. The genome of lentiviruses contains two such cis-acting purine-rich sequences: the 3'PPT common to all retroviruses and an additional central PPT present in the coding sequence of the integrase. The resulting initiation of (+) strand synthesis at the cPPT as well as at the 3'PPT leads to a (+) strand discontinuity at the centre of lentiviral genomes [1–5].
Similar to central (+) strand initiation, the termination of reverse transcription is a further feature that distinguishes lentiviruses from other orthoretroviruses. For most retroviruses, termination occurs when the RT reaches the 5' end of the template. In the case of lentiviruses however, such as HIV-1 [6], EIAV [4], and FIV [5], reverse transcription terminates ca. 100 nt downstream of the cPPT at stretches of A and T nucleotides (the central termination sequence, CTS) whose conformation disfavours binding to RT enzyme and thus halts reverse transcription. As a result of the cPPT and CTS cis-acting sequences, the final product of lentiviral reverse transcription contains a ca. 100 nt overlap, or "DNA Flap", at the centre of the genome.
Mutations introduced either in the cPPT [7–9] or in the CTS [6] of HIV-1 lead to a loss of HIV-1 infectivity that is proportional to the number and positional impact of introduced mutations. The conversion of cPPT purines into pyrimidines compromises central initiation of (+) strand synthesis and thus central DNA Flap formation [7]. Similarly, the mutation of dA n -dT n tracts in the CTS leads to aberrant (+) strand termination events spread over a distance of over 500 nt downstream of the cPPT [6]. Therefore, both the cPPT and CTS are required to form a functional central DNA Flap, and HIV-1 infection is impaired if either are mutated.
The defect in viral replication observed for cPPT mutant viruses [7, 10, 11] is not accounted for by defects in viral DNA synthesis, integration, or virus production [9] but by a defect in nuclear import [9, 10, 12]. The importance of the central DNA Flap for nuclear import was harnessed by the gene therapy field when it was shown that re-insertion of the cPPT and CTS sequences in lentiviral vectors significantly enhanced gene transfer efficiencies by increasing nuclear import [9, 13]. It is now systematically included in lentiviral vectors.
Interestingly, central (+) strand initiation has been shown to protect the HIV-1 genome from editing by cytidine deaminases of the APOBEC3 family downstream of the cPPT sequence [14]. Protection was high closest to the cPPT and decreased progressively further downstream. APOBEC3 enzymes edit exclusively single-stranded DNA, which in the case of HIV corresponds to the newly-synthesised (-) strand DNA. Editing is possible only until synthesis of the complementary (+) strand. Therefore, sequences immediately downstream of (+) strand initiation PPT sequences are more protected from editing than those upstream or further downstream [14, 15]. It has been suggested that APOBEC3G/F editing wholly accounts for the infectivity defect of cPPT mutant HIV [16]. However, the benefit of the central DNA Flap is not contested in the context of HIV-1-derived vectors, which are standardly produced in APOBEC3G-negative 293T cells [5, 9, 10, 12, 13, 17–34]. Furthermore, in the case of CTS mutants, a (+) strand segment is correctly initiated at the cPPT [6] and should protect downstream sequences from deamination as in wild-type virus. Therefore, the observed strong replication defect of CTS mutants in Vif non-permissive cells [16] cannot be attributed to APOBEC3G restriction.
Nevertheless, the importance of the central DNA Flap for HIV-1 nuclear import, infectivity and replication in vivo has been a source of much debate over the past 10 years. Some papers have proposed that the central DNA Flap contributes minimally, if at all, to HIV-1 replication, although some degree of impairment for DNA Flap mutants is observed in human PBMCs and macrophages [16, 35–38]. However, other studies claim that the central DNA Flap plays an important role in HIV nuclear import, infectivity, transduction efficiency, and viral replication [6, 7, 9–13, 17–34, 39]. Therefore, the debate seems really to reside in whether the central DNA Flap is essential for infection and replication, or whether it merely contributes towards rendering these more efficient.
Mutations in the cPPT do not lead to totally non-infectious viruses [6, 7] and some residual HIV-1 nuclear import is observed [10], suggesting either that viruses may enter the nucleus in a DNA-Flap-independent manner or that the mutations introduced in the cPPT are not sufficient to abolish DNA Flap formation. Unfortunately, the extent of mutations that can be introduced in the cPPT is limited by the need to maintain a functional integrase. The 225T mutant [17], which contains 6 synonymous mutations, maintains residual infectivity probably because the cPPT is only partially inactivated. To more profoundly disrupt DNA Flap formation, the cPPT-D mutant was generated to contain an additional K→R amino acid change and compared with a control virus cPPT-AG containing the same K→R mutation, but maintaining the polypurine nature of the cPPT [9]. Despite non-detectable (+) strand discontinuity, the cPPT-D mutant still retained residual infectivity and nuclear import (5-10% of wild-type HIV-1) [9]. Therefore, it remains an open question whether observed residual infection is due to only partial disruption of the DNA Flap, or whether a DNA-Flap-independent nuclear import exists in ca. 10% of infectious events. Since we cannot introduce any further mutations in the cPPT, we set out to combine cPPT mutations with the previously described CTS mutant that leads to aberrant (+) strand termination [6], on the premise that residual central strand initiation will then not lead to functional DNA Flap formation. We thus report the generation of fully disruptive mutants for DNA Flap formation that preserve integrase function, and document their effect on nuclear import, infectivity and replication.
In HIV-1, the cPPT sequence is an exact copy of the 3'PPT and is composed sequentially of a T-box, two A-boxes and a G-box (Figure 1). We previously found that the two A-boxes have the most profound impact on HIV-1 infectivity when mutated, followed in importance by the G- and lastly the T-box (P. Charneau, unpublished data). The 223 mutant, which introduces two pyrimidines in the A-boxes, is one of the least affected mutants [7]. The 225T mutant combines mutations in all four boxes and is the most affected amongst the synonymous cPPT mutants [17]. The addition of a single non-synonymous mutation (cPPT-D mutant) has the most profound effect on viral replication [9]. Although this mutant introduces an amino acid change at position 188 of the integrase coding region (K188R), the control virus (cPPT-AG) with the same amino acid change behaved like wild-type virus in terms of virus production and viral DNA synthesis [9]. Position 188 of integrase (IN) is important for salt bridge formation between the catalytic core and the N-terminal domain, however, the presence of an arginine rather than a lysine at this position is naturally found in HIV-2 IN and does not perturb salt bridge formation [40]. The previously described CTS mutant, which was designed to disrupt the dA n -dT n tracts of the CTS while respecting the corresponding integrase amino acid sequence, results in DNA Flaps of abnormal length and severely impairs infectivity [6].

We generated double mutants (DM) by combining in HIV-1LAI and HIV-1LAI Δenv-vsvG viruses, the 223, 225T or cPPT-D cPPT mutants with the previously described CTS mutation [6]. There were no appreciable differences in virus p24 concentration between the wild-type virus and any of the mutant viruses (Additional File 1 Figure S1), confirming that DNA Flap mutations do not affect virus production [9].
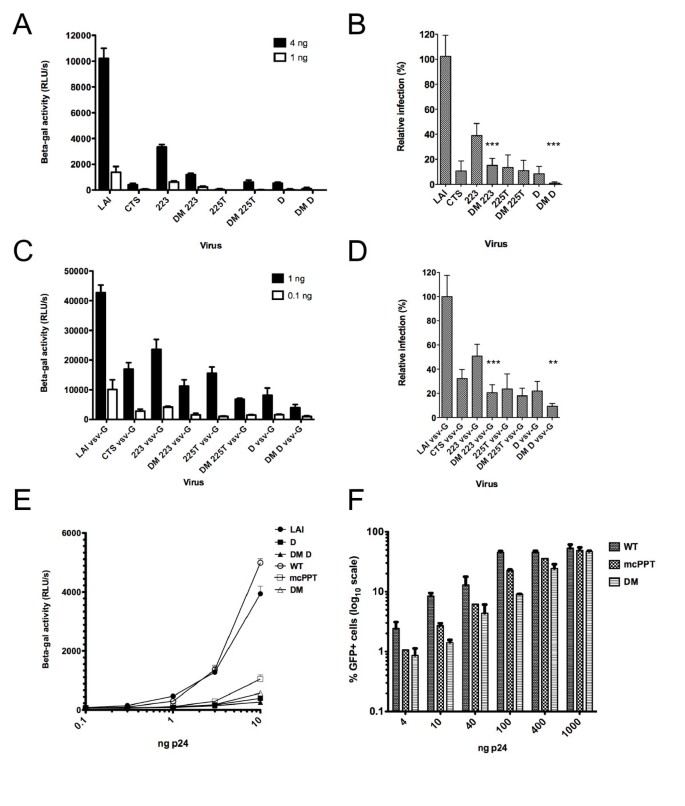
We first tested the single and double mutant viruses for infectivity in single cycle infections in the P4-CCR5 reporter cell line, which are HeLa CD4+ CXCR4+ CCR5+ carrying the Lac Z gene under the control of the HIV-1 LTR promoter [6]. cPPT and CTS HIV-1 mutants exhibited an impairment of infection that was significant and increasingly important with increasing number of mutations (Figure 2A). Reduction was consistent and was of 2-, 14-, 16- and 20-fold for 223, CTS, cPPT-D and 225T, respectively, compared to wild-type virus (Figure 2B). The combination of cPPT and CTS mutations increased this defect significantly in the case of the DM-223 and DM-D mutants. The DM-D mutant was the most affected of all DNA Flap mutants and only retained a very residual infectivity of 0.8 +/- 0.3% of wild-type virus (Figure 2B). The pseudotyping of HIV-1 with the vesicular stomatitis virus glycoprotein (VSV-G), which confers broad tropism and increases infectivity by 10-fold, attenuated the infectivity defects observed with wild-type envelope HIV-1LAI viruses (Figure 2C). The average reduction in infectivity was between 2- and 5-fold for all single mutants. However, the combining of cPPT and CTS mutations significantly further decreased the infectivity of pseudotyped viruses. The most affected mutant, DM-D VSV-G, retained an infectivity of 9.4 +/- 0.9% of wild-type virus (Figure 2D).
Iglesias, C., Ringeard, M., Di Nunzio, F. et al. Residual HIV-1 DNA Flap-independent nuclear import of cPPT/CTS double mutant viruses does not support spreading infection. Retrovirology 8, 92 (2011). https://doi.org/10.1186/1742-4690-8-92
